RdRP OTU system
PALMdb database with previously known plus Serratus OTUs palmdb repo on GitHub
Reference OTUs used in Serratus data analysis https://serratus-public.s3.amazonaws.com/rce/rdrp_otus/2021-01-25/otus-2021-01-25.tar.gz
We need a hierarchical classification of RdRPs to organize known Riboviruses and characterize new RdRPs discovered by Serratus. Taxonomy is not adequate because many known viruses are not classified at lower ranks ("unclassified Ribovirus", "unclassified Betacoronavirus" etc.), and in general sequence similarity is not sufficient to make a taxonomic assignment of a new sequence to a named clade.
Identifying a set of "known" RdRPs is not straightforward because there is no curated RdRP database analogous to SILVA for 16S and it is not trivial to extract RdRPs from GenBank due to inconsistent terminology and missing annotations. In practice, we must use sequence-based searches (diamond, motifator...) to identify RdRPs in GenBank from full-length nucleotide genomes and in translated viral ORFs and CDSs. With sequence search, there is a continuous spectrum from high confidence to low confidence and below; this must be addressed by setting thresholds which are necessarily somewhat arbitrary.
If a sequence is present only in a segment of a viral genome which is not annotated but is a high-confidence-RdRP according to motifator, is it a "known" RdRP? This could be argued both ways. For claims in a paper, we must be conservative so we will not claim it as novel. There is a spectrum from "clearly known" through "arguable" to "clearly novel assuming comprehensive due diligence in finding RdRPs in GenBank".
We should make our best effort to collect high-confidence RdRPs ("gold") and perhaps also lower-confidence RdRPs ("bronze", because silver sounds too good).
Gold sequences will be the foundation for classifying assembled RdRPs.
The "bronze" bin could contain RdRPs which are not known with high-confidence, but are identifiable easily enough that we should not claim them as novel. However, this is not needed if we use high-confidence motifator to search both known sequences and assemblies. This is because a high-confidence assembly match will be in the gold set if it is already known; it cannot be 90% similar to an RdRP in the bronze set because then motifator would have identified it with high confidence.
Possible methods include PFAM HMMs, diamond against a trusted reference, motifator, and Raffy (a random forest classifier combining inputs from diamond and motifator).
With HMMs and diamond, there are a few drawbacks. E-value is a measure of sequence similarity, but sequence similarity does not necessarily imply homology, and homology does not necessarily imply RdRP function (e.g., RTs are homologs with a different function). With diamond, we have seen that E-value < 1e-6 is a good classifier but for HMMs we haven't investigated this systematically. Also, the PFAM domains have varying overlaps with the palm segment that we want to use as a marker.
Raffy looks to work well, but it's more complicated and more difficult to use than motifator, and I see no compelling advantage to using it.
Motifator has excellent specificity for RdRP, good sensitivity, and reliably locates the palm segment boundaries. Therefore, motifator is used as the automated method for identifying RdRPs in uncurated sequences.
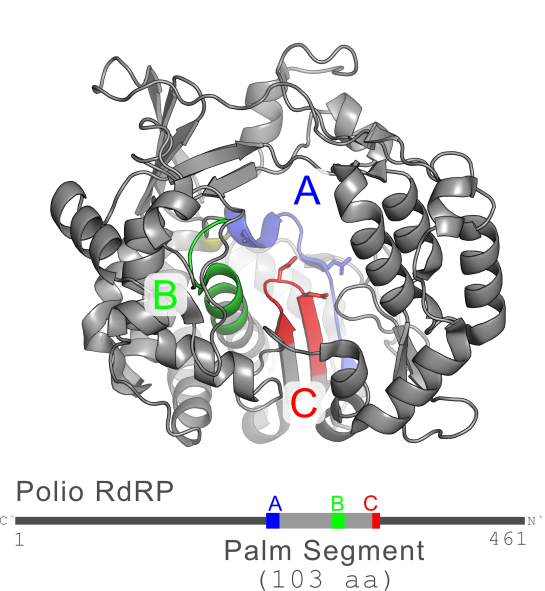
The "palm segment" is the minimal segment containing all three motifs A, B and C forming the structurally conserved catalytic core of the enzyme. Typically, the linear order is ABC in which case the palm segment extends from the first letter in A to the last letter in C. If the domain is permuted to CAB, then the segment extends from the first letter in C to the last letter in B; similarly for other permutations.
Possible methods include glocal alignment to a pre-trimmed reference such as the Wolf original sequences (before permutation hacking), and Motifator. Glocal has the problem that permutations must be identified, otherwise the trim boundaries may be quite wrong, e.g. when the closest known species has a different order of motifs. Motifator is therefore preferred as the automated method.
Manual trimming can be used in anomalous cases such as unusual permuated domains where motifator fails.
The gold RdRP set our best set of high-confidence knowns, trimmed to the palm segment. An RdRP fragment missing one or more motifs is operationally considered to be unknown. A claim of novelty is reasonable if we find the ABC segment for such a fragment. Each gold sequence is distinct; if duplicates are found in different sources these are noted in an annotation file.
The sources for gold are described next. There is no priority ordering for clustering because representative sequences are assigned new labels which are not based on database accessions. If the original accession(s) for a given unique sequence are required, then can be determined from the accession table. However, sources are treated differently depending on whether or not they are considered to be curated sets of RdRPs.
Proteins annotated by UniProt as RdRP. These are assumed to be curated; any sequence predicted as RdRP with low- or high-confidence by motifator is included.
The current Serratus query from AB. I am assuming this was monkey-curated; all motifator hits are included.
RdRPs from Koonin paper (from AB). Considered to be curated; all motifator hits are included.
This is a complete (?) set of viral ORFs from GenBank from AB. I don't know exactly how it was built. From these, only high-confidence motifator predictions are included.
This is a complete (?) set of viral nucleotide sequences downloaded from GenBank by AB. From these, only high-confidence motifator hits are included.
Not yet constructed, and might not be needed. These could include low-confidence RdRPs from gb241 plus other non-curated sources.
Gold is constructed from the above sources, giving a set of unique trimed RdRP sequences. OTUs are constructed from these by UCLUST at species-like, genus-like and family-like thresholds (90%, 75% and 40%). At each stage (uniques, 90, 75, 40), input to UCLUST is the centroids from the previous stage.
Not currently documented, mostly self-explanatory.
Once an OTU system is in production, it should be stable so that a given query sequence will be assigned to the same OTU. To maintain a stable reference, new gold sequences will be added as follows. All new sequences that do not match otus.fa at 90% identity are collected in new.fa. The unique sequences in new.fa are identified as new.uniques.fa; each of these receives a new unique integer identifier (u). These are clustered at 90% identity, giving centroids new.id90.fa which are assigned new species-like identifiers (s). Then, new.id90.fa is clustered at 75% giving centroids new.id75.fa with new g identifiers, and new.id75.fa is clustered at 40% giving centroids new.id40.fa each of which is assigned a new f identifier.
Sequences with more than ten Xs are discarded. We need to keep sequences with Xs in the gold set because otherwise we might claim a novel RdRP which is actually known except for a few Xs.