PlotHiC is used to visualize whole genome-wide contact heatmaps after genome scaffolding.
If you have any questions, please Open Issues or provide us with your comments via the email below.
Email: [email protected]
PlotHiC is utilised for the purpose of visualising genome-wide interaction heatmaps subsequent to de novo genome assembly. The software is compatible with both .hic and bed formats, and it is capable of adding chromosome names and custom visualization areas.
- Dependency :
python = "^3.10"
# pip install
pip install plothic
# create plothic enviorment and install plothic
conda env create -n plothic -c bioconda plothic
# mamba env create -n plothic -c bioconda plothicIf you want to see detailed usage and documentation of plothic, you can get it from wiki.
A simple example of PlotHiC use .hic(from Juicer/3D-DNA)and bed(from HiCPro)format is presented below.
- Input file:
genome.hic
This file is taken directly from 3d-dna, you need to select the final hic file (which has already been error adjusted and chromosome boundaries determined).
- Input file:
chr.tx(3 columns as follows, use "\t" as separator)
- This file is used for heatmap labeling. The first column is the name of the chromosome.
- The second column is the length of the chromosome (this length is the length of the hic file in Juicebox and can be manually determined from Juicebox).
- The third column is the order in which the chromosomes are placed, which is used to customize the arrangement of chromosomes (for example, from max to min).
Note: the length is in .hic file, not true base length (example as below).
# name length index
Chr1 24800000 5
Chr2 44380000 4
Chr3 63338000 3
Chr4 81187000 2
Chr5 97650000 1- Example
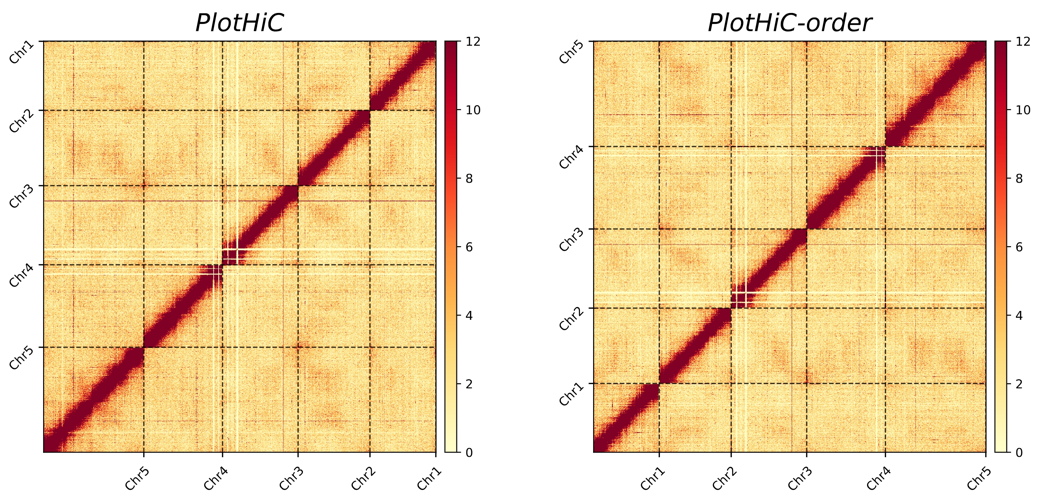
# Default order (left)
plothic -hic genome.hic -chr chr.txt -r 100000
# -hic > .hic file
# -chr > chromosome length (in .hic file)
# -r > resolution to visualization
# Custom order (right )
plothic -hic genome.hic -chr chr.txt -r 100000 --order
# --order > Sort by the order in chr.txt(index)
If the color performance is not to your liking, you can set parameters --bar-max to adjust it, which is very useful.
- Input file:
genome.matrix - Input file:
genome_abs.bed
Both files are output from HiCPro. You can select a specific resolution based on your needs.
- Input file (optional):
order.txt, the content and format as follows:
# chr_name order
NC_003070.9 1
NC_003071.7 4
NC_003074.8 3
NC_003075.7 5
NC_003076.8 2- Example
# Default order (left)
plothic -matrix sample_500000.matrix --abs-bed sample_500000_abs.bed -format png -cmap viridis --bar-max 10000 -g PlotHiC
# -matrix > matrix file
# --abs-bed > abs bed file
# -format > result format
# -cmap > viridis color
# --bar-max > max contact to show
# -g > Genome name to show top
# Custom order (right )
plothic -matrix sample_500000.matrix --abs-bed sample_500000_abs.bed -format png -cmap viridis --bar-max 10000 -g PlotHiC-order --abs-order order.txt
# --abs-order > Sort by the order in order.txt
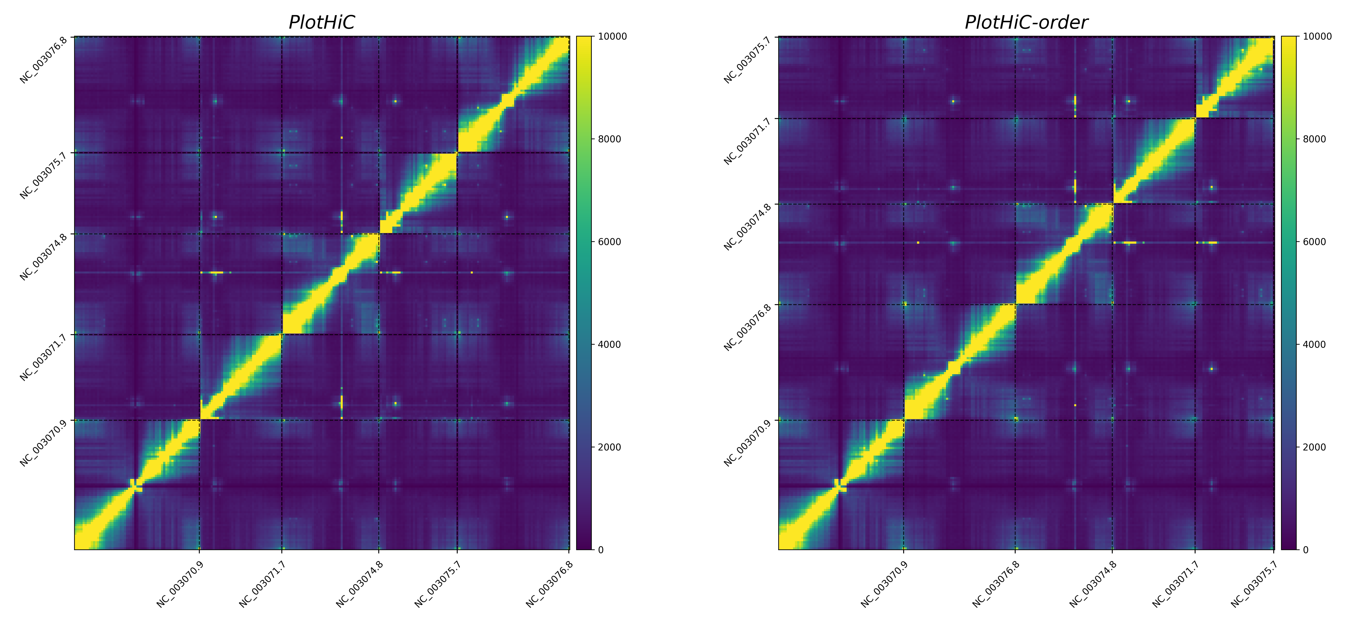

PlotHiC uses YlOrRd by default, you can choose more colors from Matplotlib.
If you used PlotHiC in your research, please cite us:
Zijie Jiang, Zhixiang Peng, Zhaoyuan Wei, Jiahe Sun, Yongjiang Luo, Lingzi Bie, Guoqing Zhang, Yi Wang, A deep learning-based method enables the automatic and accurate assembly of chromosome-level genomes, Nucleic Acids Research, 2024;, gkae789, https://doi.org/10.1093/nar/gkae789