The main goal of the fuc package (pronounced "eff-you-see") is to wrap some of the most frequently used commands in the field of bioinformatics into one place.
The package is written in Python, and supports both command line interface (CLI) and application programming interface (API) whose documentations are available at the Read the Docs.
Currently, fuc can be used to analyze, summarize, visualize, and manipulate the following file formats:
- Sequence Alignment/Map (SAM)
- Binary Alignment/Map (BAM)
- CRAM
- Variant Call Format (VCF)
- Mutation Annotation Format (MAF)
- Browser Extensible Data (BED)
- FASTQ
- FASTA
- General Feature Format (GFF)
- Gene Transfer Format (GTF)
- delimiter-separated values format (e.g. comma-separated values or CSV format)
Additionally, fuc can be used to parse output data from the following programs:
Your contributions (e.g. feature ideas, pull requests) are most welcome.
If you use fuc in a published analysis, please report the program version and cite the following article:
Lee et al., 2022. ClinPharmSeq: A targeted sequencing panel for clinical pharmacogenetics implementation. PLOS ONE.
If you find my work useful, please consider becoming a sponsor.
The following packages are required to run fuc:
biopython lxml matplotlib matplotlib-venn numpy pandas pyranges pysam scipy seaborn statsmodels
There are various ways you can install fuc. The recommended way is via conda (Anaconda):
$ conda install -c bioconda fuc
Above will automatically download and install all the dependencies as well. Alternatively, you can use pip (PyPI) to install fuc and all of its dependencies:
$ pip install fuc
Finally, you can clone the GitHub repository and then install fuc locally:
$ git clone https://github.com/sbslee/fuc
$ cd fuc
$ pip install .
The nice thing about this approach is that you will have access to development versions that are not available in Anaconda or PyPI. For example, you can access a development branch with the git checkout command. When you do this, please make sure your environment already has all the dependencies installed.
For detailed documentations on the fuc package's CLI and API, please refer to the Read the Docs.
For getting help on the fuc CLI:
$ fuc -h
usage: fuc [-h] [-v] COMMAND ...
positional arguments:
COMMAND
bam-aldepth Compute allelic depth from a BAM file.
bam-depth Compute per-base read depth from BAM files.
bam-head Print the header of a BAM file.
bam-index Index a BAM file.
bam-rename Rename the sample in a BAM file.
bam-slice Slice a BAM file.
bed-intxn Find the intersection of BED files.
bed-sum Summarize a BED file.
cov-concat Concatenate depth of coverage files.
cov-rename Rename the samples in a depth of coverage file.
fa-filter Filter sequence records in a FASTA file.
fq-count Count sequence reads in FASTQ files.
fq-sum Summarize a FASTQ file.
fuc-bgzip Write a BGZF compressed file.
fuc-compf Compare the contents of two files.
fuc-demux Parse the Reports directory from bcl2fastq.
fuc-exist Check whether certain files exist.
fuc-find Retrieve absolute paths of files whose name matches a
specified pattern, optionally recursively.
fuc-undetm Compute top unknown barcodes using undertermined FASTQ from
bcl2fastq.
maf-maf2vcf Convert a MAF file to a VCF file.
maf-oncoplt Create an oncoplot with a MAF file.
maf-sumplt Create a summary plot with a MAF file.
maf-vcf2maf Convert a VCF file to a MAF file.
ngs-bam2fq Pipeline for converting BAM files to FASTQ files.
ngs-fq2bam Pipeline for converting FASTQ files to analysis-ready BAM
files.
ngs-hc Pipeline for germline short variant discovery.
ngs-m2 Pipeline for somatic short variant discovery.
ngs-pon Pipeline for constructing a panel of normals (PoN).
ngs-quant Pipeline for running RNAseq quantification from FASTQ files
with Kallisto.
ngs-trim Pipeline for trimming adapters from FASTQ files.
tabix-index Index a GFF/BED/SAM/VCF file with Tabix.
tabix-slice Slice a GFF/BED/SAM/VCF file with Tabix.
tbl-merge Merge two table files.
tbl-sum Summarize a table file.
vcf-call Call SNVs and indels from BAM files.
vcf-filter Filter a VCF file.
vcf-index Index a VCF file.
vcf-merge Merge two or more VCF files.
vcf-rename Rename the samples in a VCF file.
vcf-slice Slice a VCF file for specified regions.
vcf-split Split a VCF file by individual.
vcf-vcf2bed Convert a VCF file to a BED file.
vcf-vep Filter a VCF file by annotations from Ensembl VEP.
optional arguments:
-h, --help Show this help message and exit.
-v, --version Show the version number and exit.
For getting help on a specific command (e.g. vcf-merge):
$ fuc vcf-merge -h
Below is the list of submodules available in the fuc API:
- common : The common submodule is used by other fuc submodules such as pyvcf and pybed. It also provides many day-to-day actions used in the field of bioinformatics.
- pybam : The pybam submodule is designed for working with sequence alignment files (SAM/BAM/CRAM). It essentially wraps the pysam package to allow fast computation and easy manipulation. If you are mainly interested in working with depth of coverage data, please check out the pycov submodule which is specifically designed for the task.
- pybed : The pybed submodule is designed for working with BED files. It implements
pybed.BedFramewhich stores BED data aspandas.DataFramevia the pyranges package to allow fast computation and easy manipulation. The submodule strictly adheres to the standard BED specification. - pychip : The pychip submodule is designed for working with annotation or manifest files from the Axiom (Thermo Fisher Scientific) and Infinium (Illumina) array platforms.
- pycov : The pycov submodule is designed for working with depth of coverage data from sequence alingment files (SAM/BAM/CRAM). It implements
pycov.CovFramewhich stores read depth data aspandas.DataFramevia the pysam package to allow fast computation and easy manipulation. Thepycov.CovFrameclass also contains many useful plotting methods such asCovFrame.plot_regionandCovFrame.plot_uniformity. - pyfq : The pyfq submodule is designed for working with FASTQ files. It implements
pyfq.FqFramewhich stores FASTQ data aspandas.DataFrameto allow fast computation and easy manipulation. - pygff : The pygff submodule is designed for working with GFF/GTF files. It implements
pygff.GffFramewhich stores GFF/GTF data aspandas.DataFrameto allow fast computation and easy manipulation. The submodule strictly adheres to the standard GFF specification. - pykallisto : The pykallisto submodule is designed for working with RNAseq quantification data from Kallisto. It implements
pykallisto.KallistoFramewhich stores Kallisto's output data aspandas.DataFrameto allow fast computation and easy manipulation. Thepykallisto.KallistoFrameclass also contains many useful plotting methods such asKallistoFrame.plot_differential_abundance. - pymaf : The pymaf submodule is designed for working with MAF files. It implements
pymaf.MafFramewhich stores MAF data aspandas.DataFrameto allow fast computation and easy manipulation. Thepymaf.MafFrameclass also contains many useful plotting methods such asMafFrame.plot_oncoplotandMafFrame.plot_summary. The submodule strictly adheres to the standard MAF specification. - pysnpeff : The pysnpeff submodule is designed for parsing VCF annotation data from the SnpEff program. It should be used with
pyvcf.VcfFrame. - pyvcf : The pyvcf submodule is designed for working with VCF files. It implements
pyvcf.VcfFramewhich stores VCF data aspandas.DataFrameto allow fast computation and easy manipulation. Thepyvcf.VcfFrameclass also contains many useful plotting methods such asVcfFrame.plot_comparisonandVcfFrame.plot_tmb. The submodule strictly adheres to the standard VCF specification. - pyvep : The pyvep submodule is designed for parsing VCF annotation data from the Ensembl VEP program. It should be used with
pyvcf.VcfFrame.
For getting help on a specific submodule (e.g. pyvcf):
>>> from fuc import pyvcf
>>> help(pyvcf)In Jupyter Notebook and Lab, you can see the documentation for a python
function by hitting SHIFT + TAB. Hit it twice to expand the view.
SAM/BAM/CRAM
To print the header of a SAM file:
$ fuc bam-head in.sam
To index a CRAM file:
$ fuc bam-index in.cram
To rename the samples in a SAM file:
$ fuc bam-rename in.sam NA12878 > out.sam
To slice a BAM file:
$ fuc bam-slice in.bam chr1:100-200 > out.bam
BED
To find intersection between BED files:
$ fuc bed-intxn 1.bed 2.bed 3.bed > intersect.bed
FASTQ
To count sequence reads in a FASTQ file:
$ fuc fq-count example.fastq
FUC
To check whether a file exists in the operating system:
$ fuc fuc-exist example.txt
To find all VCF files within the current directory recursively:
$ fuc fuc-find .vcf.gz
TABLE
To merge two tab-delimited files:
$ fuc tbl-merge left.tsv right.tsv > merged.tsv
VCF
To merge VCF files:
$ fuc vcf-merge 1.vcf 2.vcf 3.vcf > merged.vcf
To filter a VCF file annotated by Ensembl VEP:
$ fuc vcf-vep in.vcf 'SYMBOL == "TP53"' > out.vcf
BAM
To create read depth profile of a region from a CRAM file:
>>> from fuc import pycov
>>> cf = pycov.CovFrame.from_file('HG00525.final.cram', zero=True,
... region='chr12:21161194-21239796', names=['HG00525'])
>>> cf.plot_region('chr12:21161194-21239796')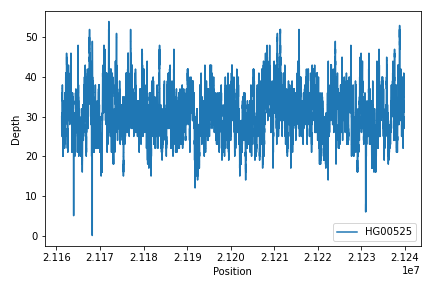
VCF
To filter a VCF file based on a BED file:
>>> from fuc import pyvcf
>>> vf = pyvcf.VcfFrame.from_file('original.vcf')
>>> filtered_vf = vf.filter_bed('targets.bed')
>>> filtered_vf.to_file('filtered.vcf')To remove indels from a VCF file:
>>> from fuc import pyvcf
>>> vf = pyvcf.VcfFrame.from_file('with_indels.vcf')
>>> filtered_vf = vf.filter_indel()
>>> filtered_vf.to_file('no_indels.vcf')To create a Venn diagram showing genotype concordance between groups:
>>> from fuc import pyvcf, common
>>> common.load_dataset('pyvcf')
>>> f = '~/fuc-data/pyvcf/plot_comparison.vcf'
>>> vf = pyvcf.VcfFrame.from_file(f)
>>> a = ['Steven_A', 'John_A', 'Sara_A']
>>> b = ['Steven_B', 'John_B', 'Sara_B']
>>> c = ['Steven_C', 'John_C', 'Sara_C']
>>> vf.plot_comparison(a, b, c)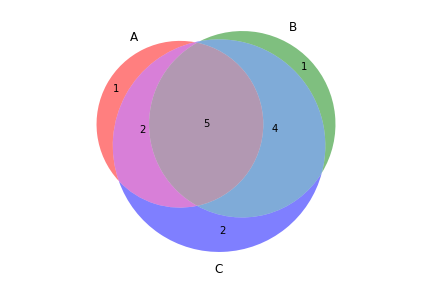
To create various figures for normal-tumor analysis:
>>> import matplotlib.pyplot as plt
>>> from fuc import common, pyvcf
>>> common.load_dataset('pyvcf')
>>> vf = pyvcf.VcfFrame.from_file('~/fuc-data/pyvcf/normal-tumor.vcf')
>>> af = pyvcf.AnnFrame.from_file('~/fuc-data/pyvcf/normal-tumor-annot.tsv', sample_col='Sample')
>>> normal = af.df[af.df.Tissue == 'Normal'].index
>>> tumor = af.df[af.df.Tissue == 'Tumor'].index
>>> fig, [[ax1, ax2], [ax3, ax4]] = plt.subplots(2, 2, figsize=(10, 10))
>>> vf.plot_tmb(ax=ax1)
>>> vf.plot_tmb(ax=ax2, af=af, group_col='Tissue')
>>> vf.plot_hist_format('#DP', ax=ax3, af=af, group_col='Tissue')
>>> vf.plot_regplot(normal, tumor, ax=ax4)
>>> plt.tight_layout()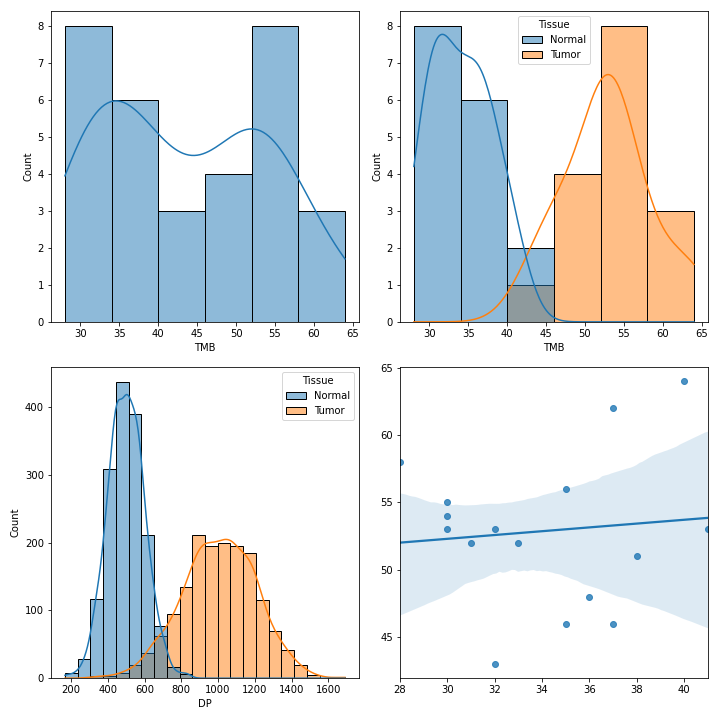
MAF
To create an oncoplot with a MAF file:
>>> from fuc import common, pymaf
>>> common.load_dataset('tcga-laml')
>>> maf_file = '~/fuc-data/tcga-laml/tcga_laml.maf.gz'
>>> mf = pymaf.MafFrame.from_file(maf_file)
>>> mf.plot_oncoplot()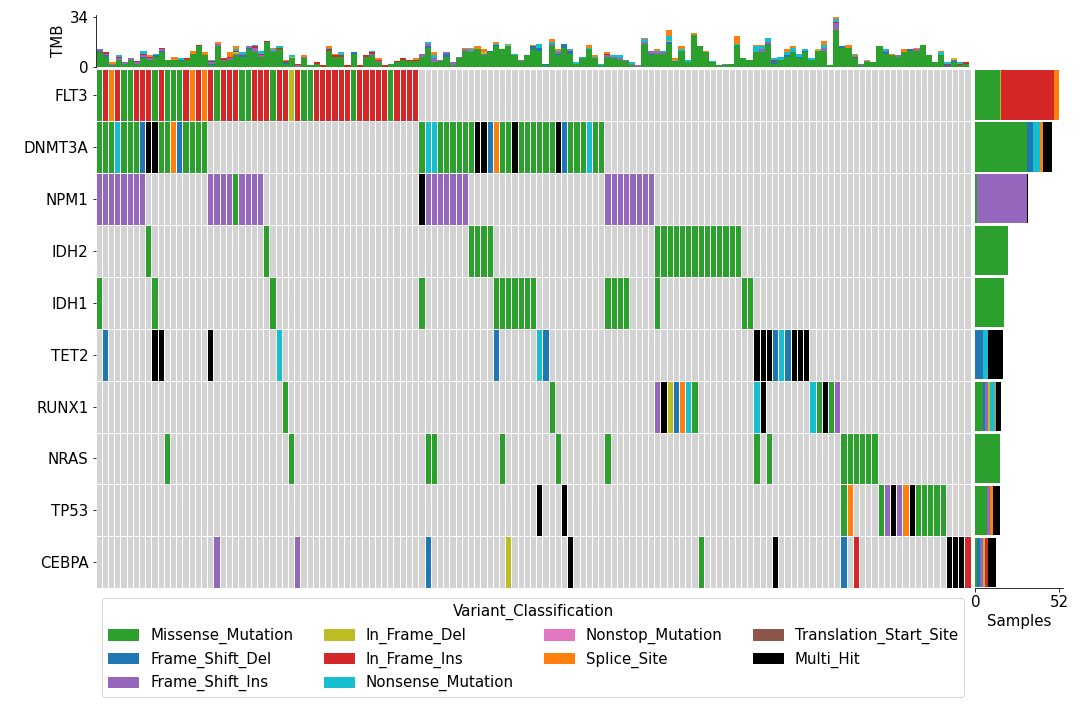
To create a customized oncoplot with a MAF file, see the Create customized oncoplot tutorial:
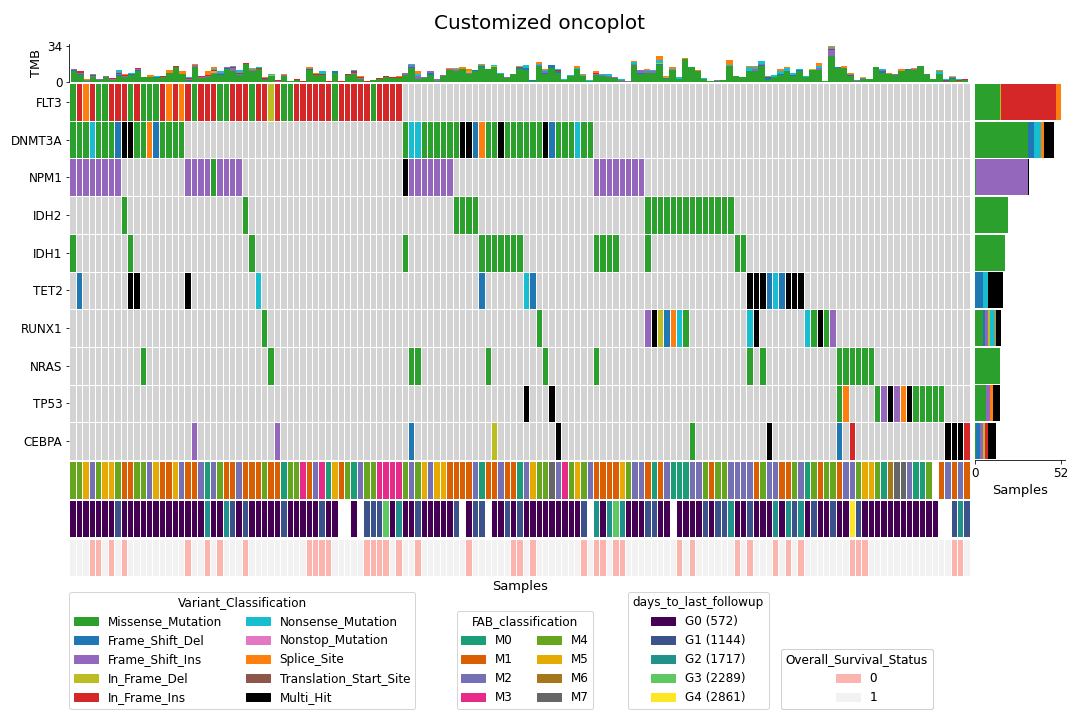
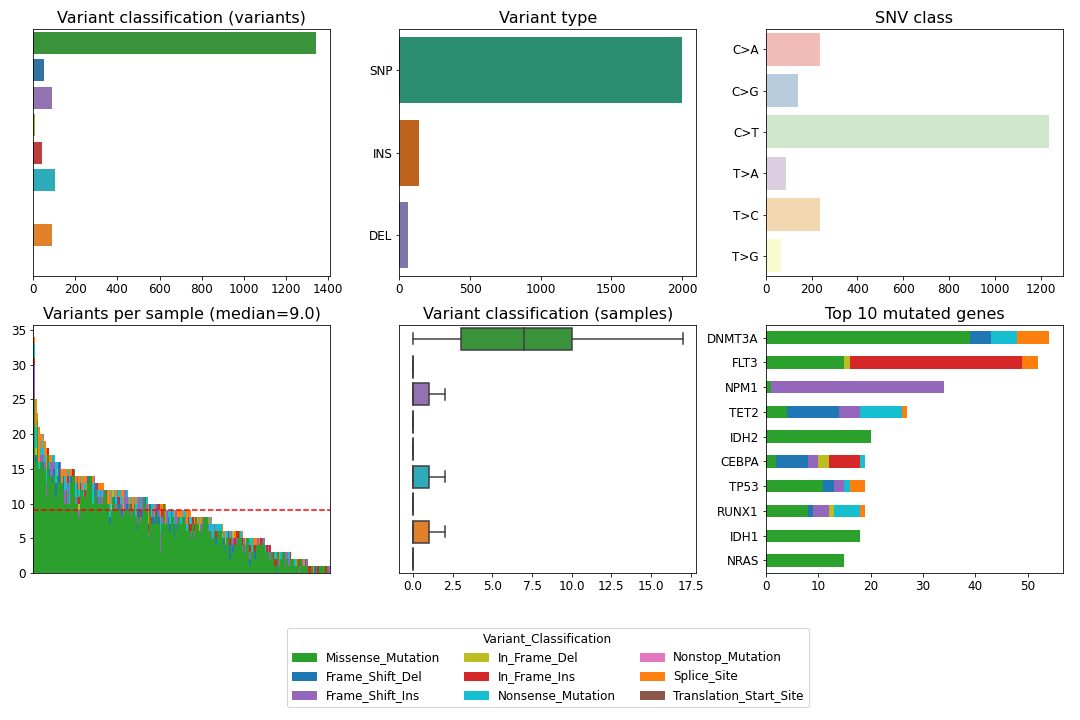
To create a summary figure for a MAF file:
>>> from fuc import common, pymaf
>>> common.load_dataset('tcga-laml')
>>> maf_file = '~/fuc-data/tcga-laml/tcga_laml.maf.gz'
>>> mf = pymaf.MafFrame.from_file(maf_file)
>>> mf.plot_summary()