MDANCE (Molecular Dynamics Analysis with N-ary Clustering Ensembles) is a flexible n-ary clustering package that provides a set of tools for clustering Molecular Dynamics trajectories. The package is written in Python and an extension of the n-ary similarity framework. The package is designed to be modular and extensible, allowing for the addition of new clustering algorithms and similarity metrics.Research contained in this package was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM150620.
$ pip install mdanceTo check for proper installation, run the following command:
>>> import mdance
>>> mdance.__version__Molecular Dynamics (MD) simulations are a powerful tool for studying the dynamics of biomolecules. However, the analysis of MD trajectories is challenging due to the large amount of data generated. Clustering is an unsupervised machine learning approach to group similar frames into clusters. The clustering results can be used to reveal the structure of the data, identify the most representative structures, and to study the dynamics of the system.
![]()
k-Means N-Ary Natural Initiation (NANI) is an algorithm for selecting initial centroids for k-Means clustering. NANI is an extension of the k-Means++ algorithm. NANI stratifies the data to high density region and perform diversity selection on top of the it to select the initial centroids. This is a deterministic algorithm that will always select the same initial centroids for the same dataset and improve on k-means++ by reducing the number of iterations required to converge and improve the clustering quality.
>>> from mdance.cluster.nani import KmeansNANI
>>> data = np.load('data.npy')
>>> N = 4
>>> mod = KmeansNANI(data, n_clusters=N, metric='MSD', N_atoms=1)
>>> initiators = mod.initiate_kmeans()
>>> initiators = initiators[:N]
>>> kmeans = KMeans(N, init=initiators, n_init=1, random_state=None)
>>> kmeans.fit(data)A tutorial is available for NANI here.
For more information on the NANI algorithm, please refer to the NANI paper.
![]()
|
Protein Retrieval via Integrative Molecular Ensembles (PRIME) is a novel algorithm that predicts the native structure of a protein from simulation or clustering data. These methods perfectly mapped all the structural motifs in the studied systems and required unprecedented linear scaling. |
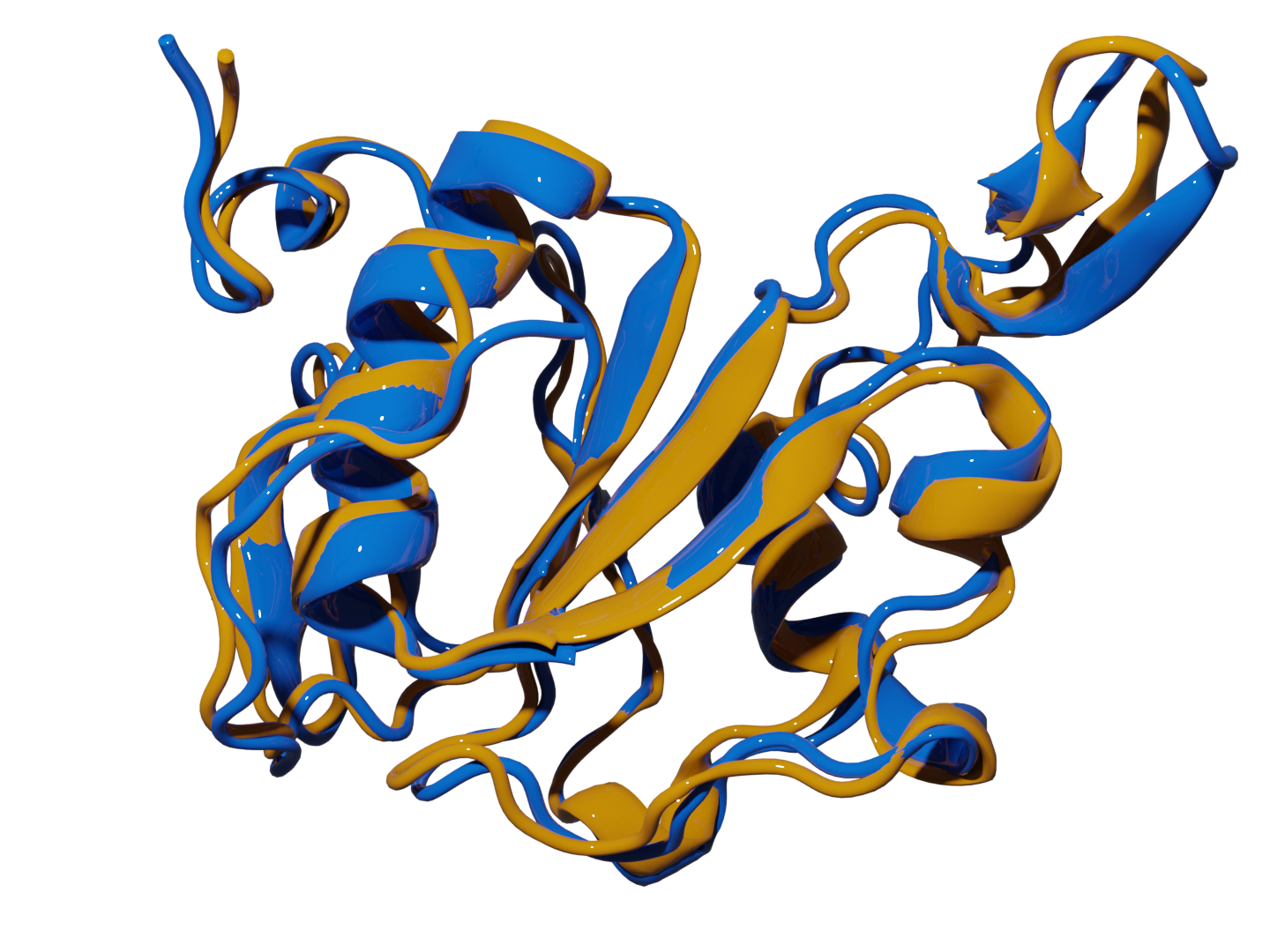 Fig 1. Superposition of the most representative structures found with extended indices (yellow) and experimental native structures (blue) of 2k2e.
Fig 1. Superposition of the most representative structures found with extended indices (yellow) and experimental native structures (blue) of 2k2e.
|
A tutorial is available for PRIME here.
For more information on the PRIME algorithm, please refer to the PRIME paper.
Please! Don't hesitate to reach out!