

A command line application to launch molecular dynamics simulations with OpenMM
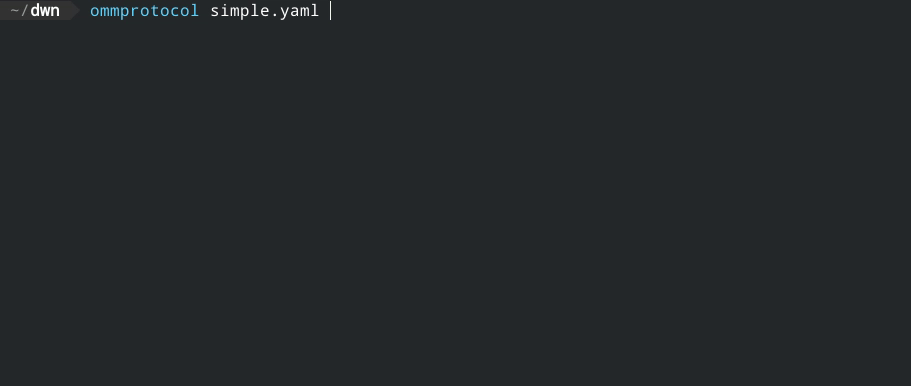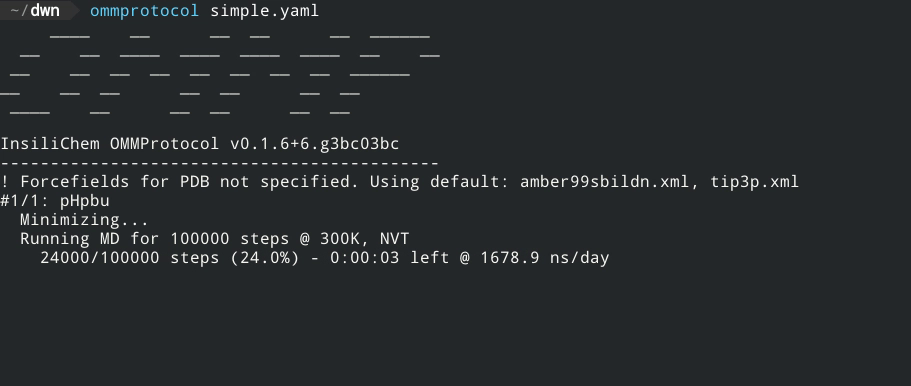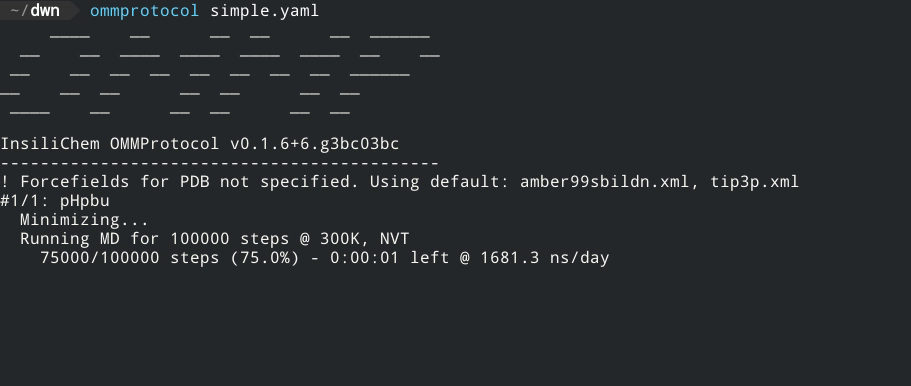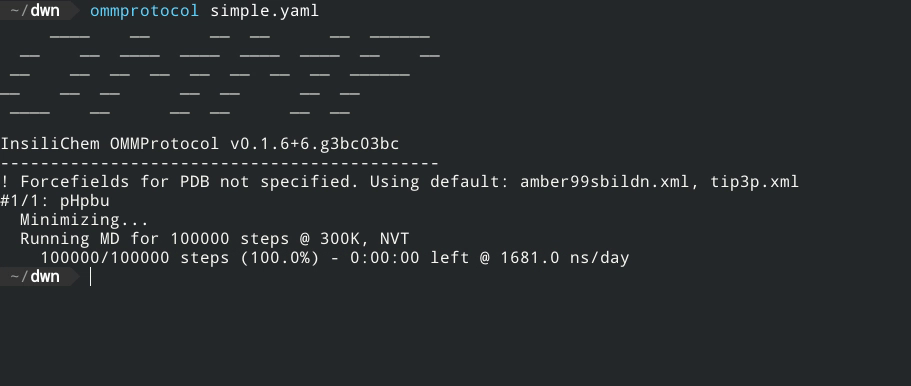
- No coding required - just a YAML input file!
- Smart support for different input file formats:
- Topology: PDB/PDBx, Mol2, Amber's PRMTOP, Charmm's PSF, Gromacs' TOP, Desmond's DMS
- Positions: PDB, COOR, INPCRD, CRD, GRO
- Velocities: PDB, VEL
- Box vectors: PDB, XSC, CSV, INPCRD, GRO
- A fallback method is implemented and will attempt to load verything else that might be supported by ParmEd.
- Choose your preferred trajectory format (PDB, PDBx, DCD, HDF5, NETCDF, MDCRD) and checkpoints (Amber's, CHARMM's, OpenMM XML states).
- Live report of simulation progress, with estimated ETA and speed.
- Checkpoint every n steps. Also, emergency rescue files are created if an error occurs.
- Autochunk the trajectories for easy handling.
Download the latest installer or use conda install -c omnia -c insilichem ommprotocol if you already have Anaconda/Miniconda installed. Further details here.
When installed, you should be able to run:
ommprotocol <inputfile.yaml>
Check the documentation to read more on how to create input files for OMMProtocol.
Documentation is always available at ReadTheDocs. If you have problems running ommprotocol, feel free to create an issue! Also, make sure to visit our main webpage at insilichem.com.
OMMProtocol is scientific software, funded by public research grants (Spanish MINECO's project CTQ2014-54071-P, Generalitat de Catalunya's project 2014SGR989 and research grant 2015FI_B00768, COST Action CM1306). If you make use of Ommprotocol in scientific publications, please cite it. It will help measure the impact of our research and future funding! A manuscript is in progress and available as a pre-print in ChemRxiv.
@article{ommprotocol,
author = {Rodríguez-Guerra Pedregal, Jaime and
Alonso-Cotchico, Lur and
Velasco-Carneros, Lorea and
Maréchal, Jean-Didier}
title = {OMMProtocol: A Command Line Application to Launch Molecular Dynamics Simulations with OpenMM},
url = {https://chemrxiv.org/articles/OMMProtocol_A_Command_Line_Application_to_Launch_Molecular_Dynamics_Simulations_with_OpenMM/7059263/1},
DOI = {10.26434/chemrxiv.7059263.v1}
publisher = {ChemRxiv},
year = {2018},
month = {Sep}
}