| tags |
|---|
ggg, ggg2024, ggg298 |
[toc]
This two hour lab will show students how to install and manage software using the conda installation system. We will give examples of installing Python and R software, and managing conda environments on remote systems.
This lesson was adopted from a lesson co-authored by Shannon Joslin for GGG 298 at UC Davis. There is also a really nice binder-based tutorial on the NIH CFDE training Web site!
Learning objectives:
- learn the basics of software installation, software dependencies, and isolation environments
- learn about conda and how to use it
- learn about conda-forge and bioconda and how to install software from them
- learn to use conda to manage R and Python installations
Other references:
- Remote Computing (August 2021) version of the conda lesson
- ANGUS 2019 lesson
- Why you need Python Environments and How to Manage Them with Conda
Outline:
There are many reasons!
It's a confusing ecosystem of operating systems (Mac OS X, many versions of Linux, Windows)
Many software has many dependencies (e.g. just consider base language -- C++, Java, Python, R, and their different versions)
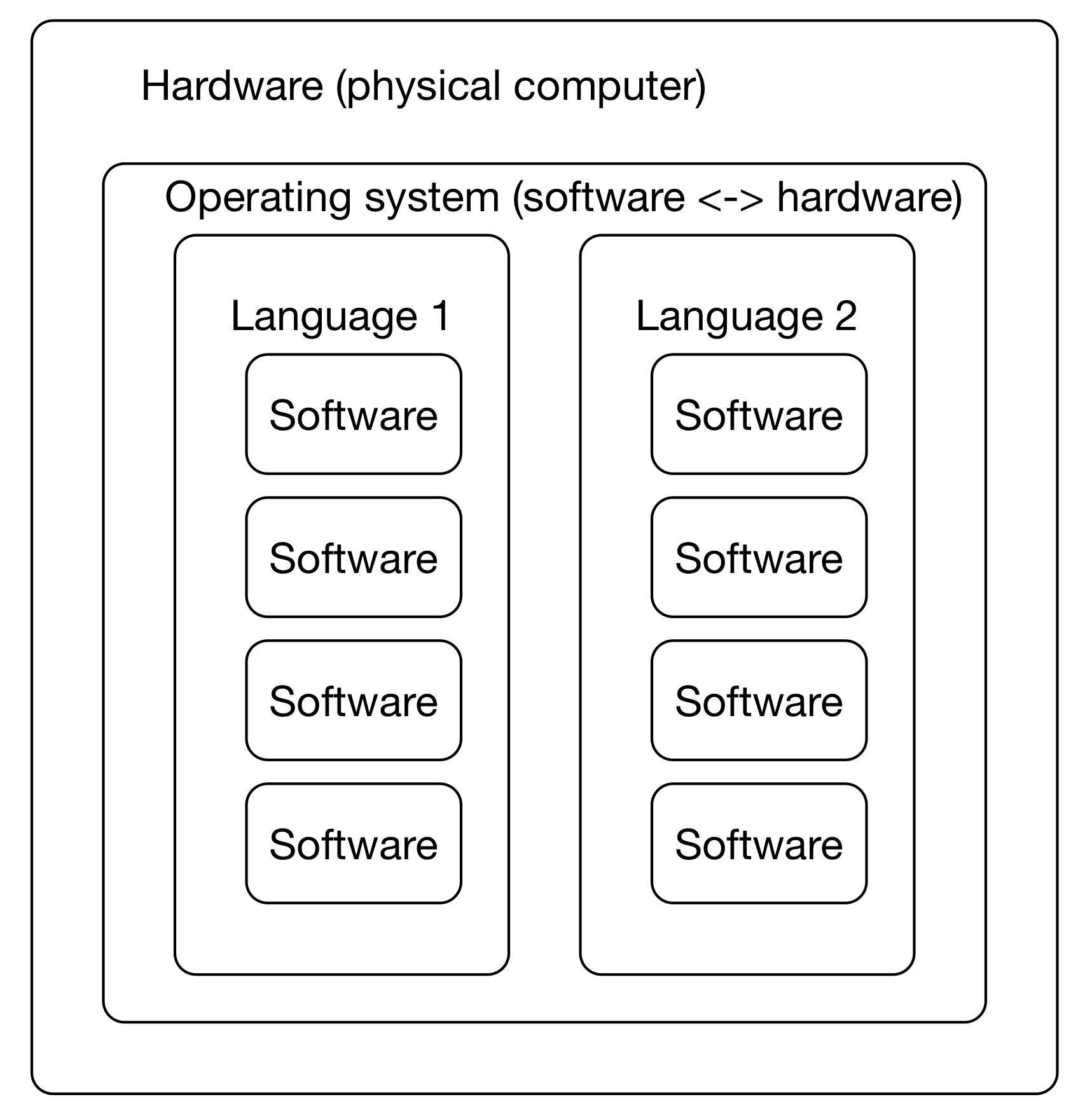
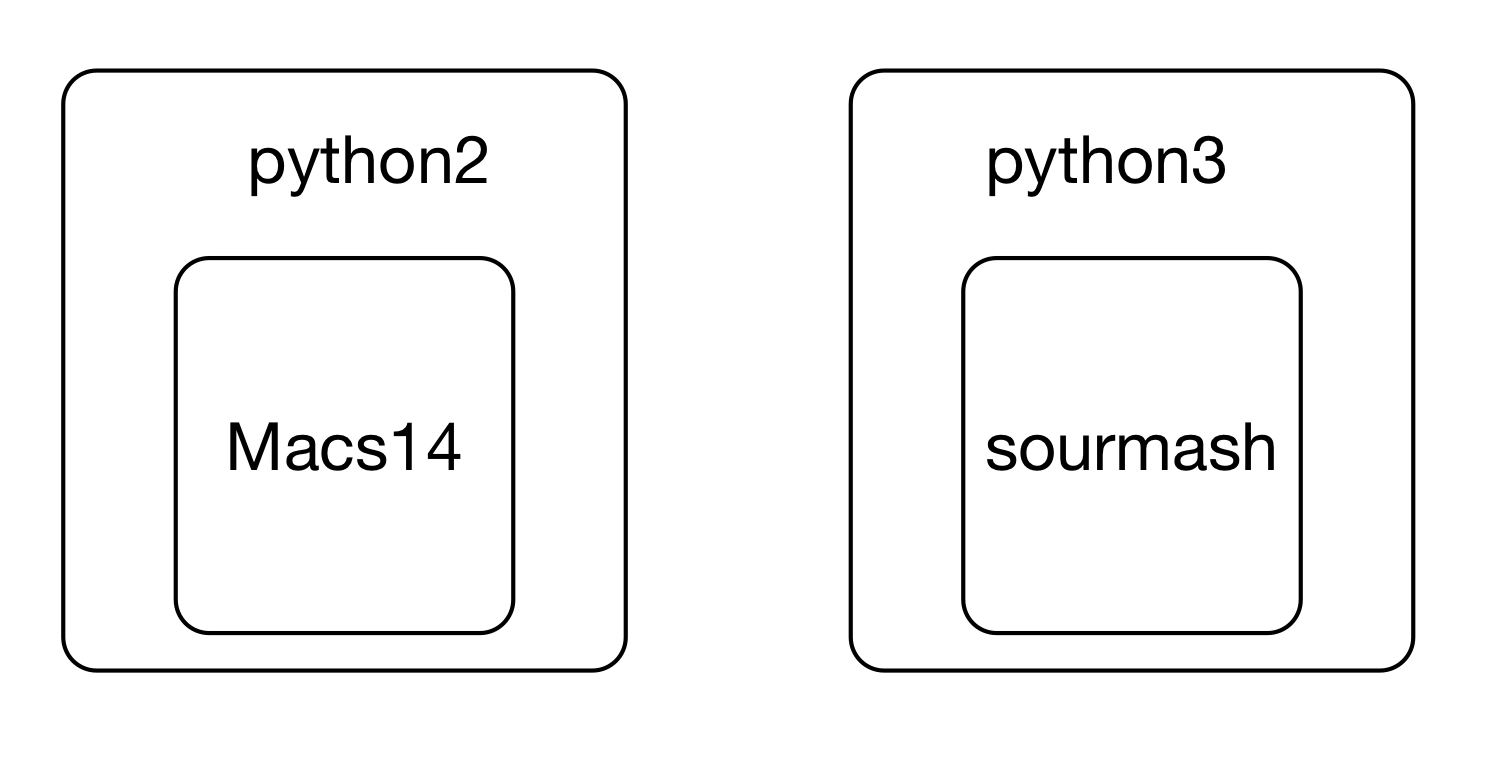
This leads to confusing situations where different versions of underlying software are need to run two different programs -- what if you wanted to run Macs14 and sourmash both, but one wanted 'python' to mean python2 and the other wanted 'python' to mean python3? Or you needed to use two R packages - one that only worked with R 4.0.1, and one that worked only with R 4.1.1?
Decoupling user-focused software from underlying operating systems is a Big Deal - imagine, otherwise you'd have to rebuild software for every OS! (This is kind of what conda does for you, actually - it's just centralized!)
Also, lot of software installation currently requires (or at least is much easier with) sysadmin privileges, which is inherently dangerous on mulituser systems.
Why do you need isolated software install environments? Some specific reasons:
- your work relies on a bunch of specific versions (perhaps old versions?)
- working with a collaborator who really likes a particular feature!
- experiment with new packages without messing up current workflow (reproducibility!)
- publication ("here's what I used for software", repeatability!)
- sometimes workflows rely on incompatible software packages! see my twitter question
Conda tries to solve all of these problems, and (in our experience) largely succeeds. That's what we'll explore today.
Note that conda emerged from the Python world but is much broader and works for many more software types, including R!
Log into farm.cse.ucdavis.edu using your datalab-XX account; see instructions.
We suggest using RStudio but you don't have to - you can run everything below at a regular ssh login prompt.
:::info If using RStudio, remember to use the Terminal tab! :::
conda/mamba is already available on the farm HPC via the modules system: type
module load mamba
and your prompt should change to look something like:
(base) datalab-05@farm:~$
The 'base' part of the prompt indicates that conda has been activated in your account and that you are in the default (base) environment. Read on!
:::warning In case you want to install it on another computer follow these instructions to install mamba!. :::
:::info
mamba is a reimplementation of conda that is faster.
You can use either conda or mamba to manage environments. They are, with almost no exceptions, identical in behavior.
Below, we'll be using the mamba command because it's always faster than conda.
:::
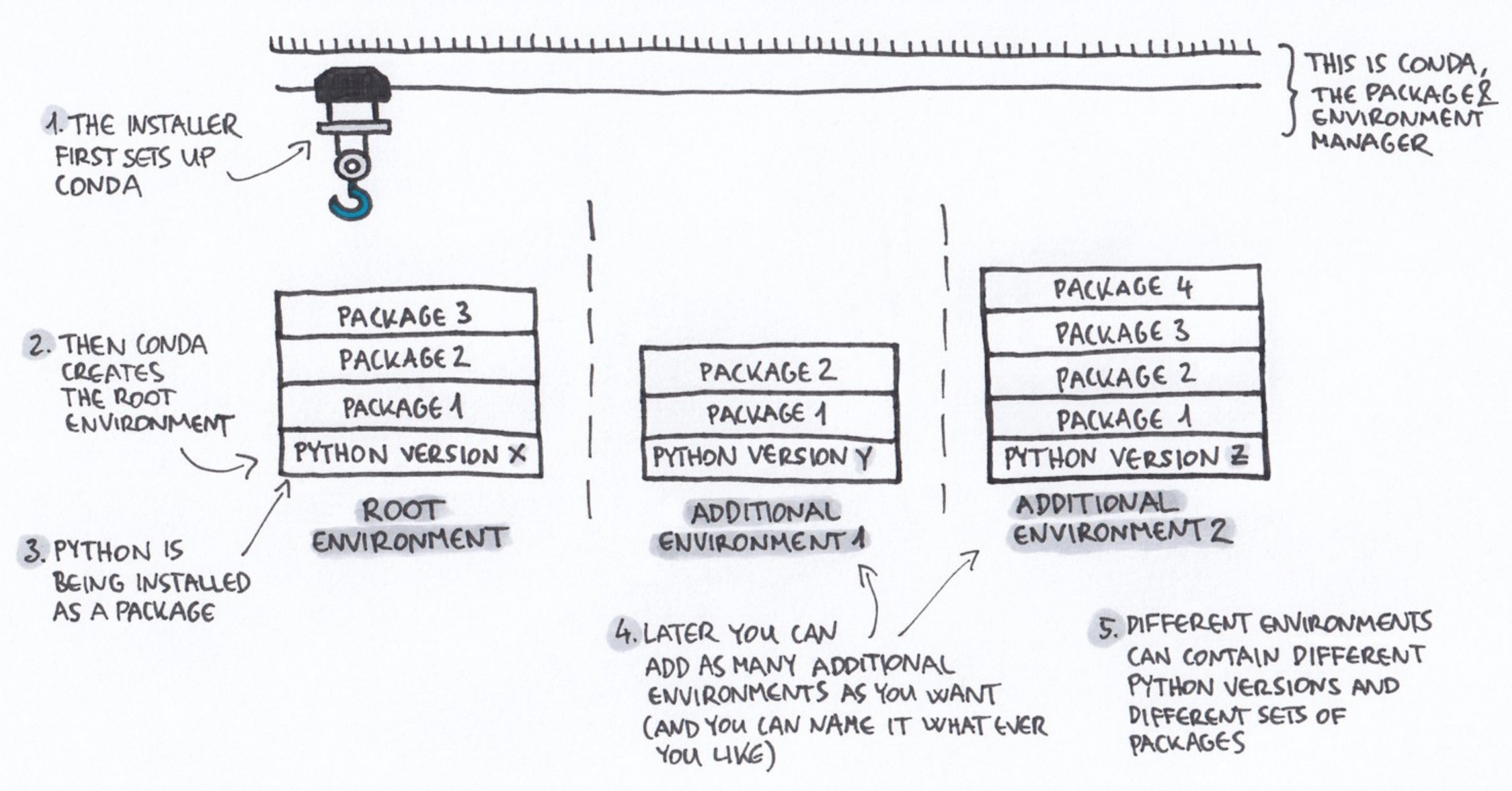
A conda environment is a specific collection of packages/tools that you have installed. For example, you might have a Python 3.11 environment for the latest version of Python, and Python 3.9 environment for an older version that works with certain packages.
Environments are
isolated from each other: if you change one environment, your other
environments are not affected. You can easily activate new
environments, which is how you switch between them.
Let's install some software! We'll start with csvtk, a tool to work with text files (see remote computing workshop 2, working with text files).
Here's the command to create a new conda environment, named 'csv', with csvtk installed.
mamba create --name csv -y csvtk
Here, we are installing the csvtk package into an environment named csv.
The -y indicates that we don't want conda to ask us for confirmation - it
should just go ahead and create the environment.
Now, activate the new environment:
mamba activate csv
Your prompt should change to have (csv) at the beginning.
Let's run csvtk on some real data now! To download the data, run:
cd ~/
git clone https://github.com/ngs-docs/2021-remote-computing-binder/
(per this workshop).
Now uncompress one of the downloaded files:
cd ~/2021-remote-computing-binder/SouthParkData
gunzip -k All-seasons.csv.gz
ls -l All-seasons.csv
You should now be able to run:
csvtk cut -f Character All-seasons.csv | grep Computer | sort | uniq -c
and you should see:
13 Computer 3 Computer Voice
What's going on here?
csvtk is a cross-platform library for working with CSV and TSV files. It's written and maintained by Wei Shen, and it's free and open source.
It's relatively new - I found out about it a few years ago - and while it doesn't do anything I couldn't do with other commands, it packages a bunch of really nice functionality together into one tool.
For example, I can find the headers in a csv file like so,
csvtk headers All-seasons.csv
and I can then cut out one particular column and summarize things -- e.g.
csvtk cut -f Character All-seasons.csv | sort | uniq -c | sort -n | tail
(which tells me that Cartman is by far the most quoted character in this file).
What I'm doing here is using UNIX piping and filtering (see more information here) to:
- take the contents of the Character column, produced by csvtk
- sorting the contents (
sort) - counting the number of distinct elements (
uniq -c) - sorting the counts from least to most by number (
sort -n) - and then showing only the last 10 (
tail)
(It looks complicated, but as you start using these commands more and more, it will become second nature!)
::::warning CHALLENGE: How would I find the least quoted characters in this file? ::::spoiler You could use either
csvtk cut -f Character All-seasons.csv | sort | uniq -c | sort -rn | tail
or
csvtk cut -f Character All-seasons.csv | sort | uniq -c | sort -n | head
::::
The key thing about csvtk for this lesson is that it's new software, and it's mostly useful for data scientists, so it's not "built into" UNIX operating systems yet (and may never be).
So when you try running it in the base environment,
mamba activate base
csvtkyou'll get "command not found" because it's not installed.
Let's revisit that:
- csvtk is not something that's installed by default on farm.
- it's not in our default conda environment, either.
- but, if we use mamba to install it in a new environment, and then activate that environment, we can use it!
If you switch back to your csv environment, you'll see that you can run it again:
conda activate csv
csvtk
When we run conda create to install csvtk, where is conda looking for
and finding the software?
The short answer is "the Internet", and more specifically, pre-prepared conda software repositories, or "channels". Bioconda is one such channel, as is conda-forge. We'll talk more about channels below.
If you're at all familiar with UNIX, you might be saying "wait! but I already use homebrew! or apt! or yum! or containers! or ...!"
Or you may point your sysadmins at this tutorial and they'll say "well you should just use the modules system on your HPC."
Yes, there are many ways to install and use software! Why conda?
We like conda, because it's straightforward, flexible, cross-platform, supports version pinning, and does not require systems administrator access. In the past 12 years of teaching bioinformatics and data science, conda has come closest to being the thing that works for everyone, on every platform. As they say, "your mileage may vary" (YMMV). But we like conda :).
More specifically,
- conda doesn't require sysadmin privileges to install stuff!
- it is not platform specific - it works on Mac OS, Linux, and Windows.
- it is open source.
- It is supported by a big community.
- Most command-line software is available via it, and that software is often up to date.
Once you're in an environment, you can install new software into that environment with conda install -y <software_name> like so:
mamba activate base
mamba install -y fastqc
and that should work too! You'll be able to run the fastqc command now.
What you're doing here is installing software into an existing environment with mamba install, instead of creating a new environment with mamba create.
:::success Let's try it out quickly -
mkdir ~/298-lab3
cd ~/298-lab3
fastqc ~ctbrown/data/ggg201b/SRR2584403_1.fastq.gz -o ./
As per our previous execution of fastqc, this will create two files, SRR2584403_1_fastqc.html and SRR2584403_1_fastqc.zip, in your 298-lab3/ directory.
:::
Generally you want to avoid installing too many packages in one environment, as it starts to get slow to figure out whether or not something can be installed. We'll talk more about this below.
You can list software with mamba list:
mamba list
which is less useful than you might think, given how many packages we've actually installed... this is because packages like FastQC have many dependencies!
(Why are there so many? Because most software is built on top of lots of other software... and so you need to install all of that other software, too!)
Installed software on most operating systems (including Linux, which is what farm runs) is usually installed in a separate place from where you run it.
You run software in a directory where you want to input or output files, e.g. your data files or results files. This directory is known as the current working directory, and it's displayed on the prompt. You can also ask what it is with pwd.
You install software into a location (directory) that's on your "path". There are lots of different things there on your path - you can look with:
echo $PATH
Some of these locations are system installation locations, some of them are module locations, and some of them are conda locations. It gets pretty complicated!
What conda activate is actually doing is adjusting your path to point at the software installed in a particular environment. For example, fastqc is installed in a directory ~/.conda/envs/csv/bin/; you can figure this out by typing:
which fastqc
These installation directories can get pretty big:
du -sh ~/.conda/envs/csv
will show you this:
529M /home/datalab-05/.conda/envs/csv
but it's still smaller than most of your data sets so 🤷
To see what version of a particular piece of software you have installed, run:
mamba list csvtk
and look at the second column. (Note that mamba list doesn't need an exact match, so e.g. you can find all packages with 'csv' in the name by doing mamba list csv).
As of Jan 2024, conda installs csvtk version 0.29.0. You can force conda to install exactly this version in the future like so,
mamba install csvtk==0.29.0
This can be really important - software versions change all the time, which may change data analysis results. Mamba lets you "pin" your software to a specific version to help control this!
:::info Unfortunately there's no good way to know if a new version of a software package is "significant" or not, unless they use semantic versioning... Generally if there's a big number update (1.0 -> 2.0) the software will work quite differently, but there's no guarantee on that.
For example, our software sourmash 1.0 was very different from 2.0, while sourmash 3.0 was virtually identical to 2.0 in usage (but not in implementation). The next version, sourmash 4.0, changed many things.
(The lesson is, it is hard for software projects to communicate changes in any systematic way!) :::
::::warning
CHALLENGE: Use the mamba create command to create a new environment (named smash)
and install the latest version of sourmash in it. Then activate that
environment and verify that you can run 'sourmash'.
::::spoiler
Try:
mamba create --name smash -y sourmash
mamba activate sourmash
sourmash
Make sure to switch back to your csv environment when you're all done:
conda activate csv
What if you want to specify collections of software that you use together? And/or send collaborators or colleagues the set of software they need, all in one nice file?
conda has a nice human-readable format for that, called an 'environment file'. These are portable files that you can put on different computers and have them automatically install the right stuff. You can see the one for the binder for a remote computing workshops here, and the one for an RNAseq pipeline here, for example.
(These are YAML files, which are in a format that's structured for computers to read but not super difficult for humans to write. You'll see these used occasionally in configuration files for bioinformatics software, too.)
If you have a complicated environment that you want to save, you can make an environment file from your current environment like so:
mamba env export > export.yml
To look at the environment files we've just created, do:
cat export.yml
and you can create a new environment from this file like so:
mamba env create -n csv2 -f export.yml
This would create a new environment called csv2 that has all the same things installed in it as csv does (because that's where we ran mamba env export!)
I should say that I don't actually recommend this, because the environment file is going to contain all of the dependencies too. I tend to just list the specific packages I care about running in the environment file; mamba will happily install all the dependencies as needed.
So, for example, an environment file for csvtk would be simply:
channels:
- conda-forge
- bioconda
- defaults
dependencies:
- csvtk
You can update software with mamba update, and remove software with
mamba remove. Generally there's not too much need for these commands
tho, as we recommend just creating new environments with a pinned
version, OR the latest version. Then, when you want to update your software,
you create a new, clean environment.
As you can probably infer from the above, you can have multiple environments with different (potentially incompatible) things installed in each one.
The default is base. Other environments will have their own set of
packages. Environments do not include packages from other environments;
you'll need to install each package in every environment that needs it.
So, for example:
mamba activate base
csvtk
should return "command not found".
:::info This may be annoying at first but it helps prevent confusion - you have to be explicit about what you want in each environment! :::
You can list environments with mamba env list:
mamba env list
It will list all of the available environments as well as denote the environment you are currently in with an *.
Switch environments with conda activate <environment_name>, and remove environments with mamba env remove -n <environment_name>.
Note that switching environments doesn't switch your working directory, it just switches the software you're running.
So, for example, you can have one working directory in which you use two different collections of software just fine; you would just have to switch mamba environments. This is really useful for workflows!
We generally recommend using a task-specific environments for each task (e.g. one environment for your R work, and another for your bioinformatics work), because this can dramatically simplify installation and speed up new conda installs. This is because conda needs to make sure that any new package (and all of its dependencies) you would like to install doesn't conflict with all the existing packages.
That having been said, my experience is that I rely on a core set of packages for my daily work, and so I usually have my "daily work" environment where I have a bunch of stuff installed, and then when I'm working on data analysis, I don't want to constantly install new things or switch environments.
So I usually have a default environment that I work in, and when I use non-standard software (stuff I use infrequently or for specific tasks) I create software-specific environments to work in.
snakemake (which we'll be talking about in more detail soon) helps with this by letting you use analysis-specific environments.
To search for all available versions of a particular package from the command line, do:
mamba search <software>
Conda-forge and Bioconda are "software channels" for conda, that provide collections of conda-packaged software.
In particular, conda-forge contains an awful lot of general community packages as well as many Python and R libraries, while bioconda is more focused on biology/bioinformatics tools specifically.
You can install stuff directly from these channels by specifying the
bioconda channel explicitly: mamba install -c bioconda .... Or, you
can add it to your "default" set of channels to search, as it is in your account (because we installed mamba for you using mambaforge).
(You don't need to run these, but you can:)
mamba config --add channels defaults
mamba config --add channels bioconda
mamba config --add channels conda-forge
These commands configure things in your .condarc file -- take a look,
cat ~/.condarc
This will automatically make mamba install search for packages in bioconda and conda-forge.
Note: if you get the error
PackagesNotFoundError: The following packages are not available from current channels:
but you're pretty sure the package exists, you probably need to configure your channels properly :)
conda and mamba work on Windows, OS X and Linux!
But unfortunately many conda-forge and bioconda packages are only available for OS X and Linux, and not Windows :(. This is because they are built for Linux or a UNIX-like OS.
Both conda-forge and bioconda are community-driven libraries of software. They rely upon people (like you!) to package software; this involves writing a recipe, like the sourmash recipe.
Some tips and tricks for using conda-forge and bioconda:
- Both are community maintained, please be respectful and friendly - no one is working for you, and no one is making money doing this.
- Help advocate for your favorite software to be added, and/or do it yourself! (e.g. gtdbtk) - we can help!
- File bug reports, but be respectful and helpful. See a sterling example :).
Conda emerged from the Python community initially, but it now has some pretty good support for R as well, through the conda-forge channel.
Why use Conda for R and Python? What does it give you? A few things -
- with conda, you can manage multiple R and Python installations, each containing different (and incompatible!) versions of packages.
- where libraries are available via conda, you can install them without compiling them. This can be (much) faster than using the standard
install.packagesorpip installsupported by R and Python. - where libraries are not available via conda, you can still install them, and you don't need to do anything special or tricky - just run the standard installation commands.
conda-forge now has some pretty good support for R - see this environment file for RNAseq, for example. It installs both tidyverse packages and bioconductor packages!
Let's install R with dplyr support.
First, let's figure out what the right package name is. We could go to the conda-forge page and search for rmarkdown, but it turns out that google is often your best bet :).
Google 'install dplyr with conda' and you'll see that the first hit is
r-dplyr. Let's try it!
mamba create -y -n dplyr r-dplyr
This will go ahead and install R itself, as well as all of the packages needed to compile RMarkdown documents (like this Web site, in fact).
Activate the environment:
conda activate r-dplyr
Now, try running:
type R
to see where R is installed - under your own account. Yay!
If you want, you can run R and then library(dplyr) to verify that
it's installed. (Type 'exit' to get out of R!)
Here's what's going on:
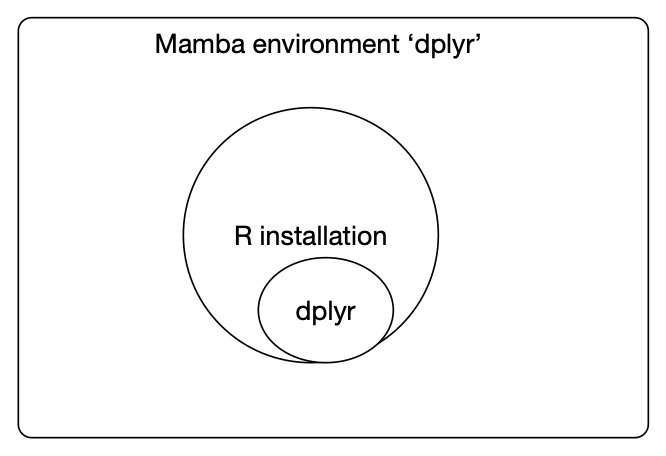
You can also install your own R packages from within R using install.packages - and, as long as you're running R from within your conda environment, it will install into that environment.
You can manage R packages either way - with conda, or "manually" using R's internal mechanisms. It's often faster to use conda, especially if there are C extension packages, and I've found that the majority of R packages I use in bioinformatics are readily available via conda-forge, which is nice. Again, your mileage may vary... regardless, at least now you have options!
You know how you enter module load R before module load rstudio-server? You can replace module load R with mamba activate dplyr, and then execute rstudio-launch, and then you will be using the R installed in the monda environment!
Python is heavily used in data science as well, and it is also well supported by conda. Conveniently, you can install different versions quite easily:
mamba create -n py311 -y python==3.11
will install Python v3.9.
Then
conda activate py311
type python
python -V
will show you that Python is v3.9 in this environment.
Conveniently, as with R, you have full installation privileges - so you could run the python package installer, pip, to install stuff. For example, run:
pip install screed
to install the screed library in your very own Python environment. (screed is another library my lab built for reading sequence data files; here we're just using it as an example of something that you can install with mamba).
Many (most?) people now use R and Python packages via RStudio and JupyterLab, and it's totally possible to use conda installs with that.
We'll cover this elsewhere (not today), but hit me up if you're interested in instructions!
This is because conda needs to make sure to resolve version incompatibilities before proceeding, and for large conda environments containing many packages, this can take a while!
This has mostly been resolved by the mamba command, which is much faster than conda.
Solution: use isolated environments to install single packages, instead.
You can grab an explicit list of version pinned software that is OS specific like so -
mamba list --explicit > package-files.txt
mamba create --name myenv --file spec-file.txt
mamba install --name myenv --file spec-file.txt
this will guarantee identical environments.
See the conda docs for more information.
| Conda commands | action |
|---|---|
conda install <package_name> |
install a package |
conda list |
list installed packages |
conda search <package_name> |
search for a package––this can be the full or partial name of a package |
conda info |
list of information about the currently active environment |
conda list |
list out all the installed packages in the currently active environment |
conda remove <package_name> |
remove a conda package |
conda config --get channels |
list out the active channels and their priorities |
conda update |
update all the installed packages |
conda config --remove channels <channel> |
remove unwanted channel |
conda env list |
list the different environments you have set up |
conda activate <new_environment_name> |
activate the a new conda environment conda (this also works for activating our base environment |
conda info --envs |
list the locations of conda directories |
- Conda Documentation
- Drawing credit: Gergely Szerovay. Read original article here
- What happens if something isn't conda installable?
- You can install it as normal, and it will be usable as normal.
- However, it won't be "managed" via conda (or snakemake)
- Disk, vs environment, vs login shell
- especially note that multiple terminals all look at the same disk
I don't yet use this myself, but I thought it would be fun to try coming at this from a different direction --
Let's use a completely different interface that's built on top of conda, called pixi! It uses all the same software packages and so on, but uses a different strategy for activating environments.
first, we need to install pixi:
curl -fsSL https://pixi.sh/install.sh | bash
and activate it:
source ~/.bashrc
cd ~/
mkdir 298-pixi
cd 298-pixi
pixi init .
pixi project channel add bioconda
pixi add fastqc
pixi run fastqc ~ctbrown/data/ggg201b/SRR2584857_1.fastq.gz -o .
What's different about pixi?
Are these differences good or bad?
It seems like you can use them both at the same time 🤷 🤯
Mamba/conda is one way you can install software on computers, including most especially HPC and cloud computers.
Mamba lets you create separate "environments" containing collections of software that are isolated from other collections.
Mamba supports a pretty normal data science set of tools, and also provides tools to support computational reproducibility via environment files and version pinning.
Go mamba!!